Wilson’s disease is an autosomal recessive condition where copper builds up in the body, particularly the liver and brain.
Epidemiology
Wilson’s disease is relatively rare, with the worldwide prevalence estimated to be 1 in 30,000. Males and females are affected equally, and patients present between the age of 4 and 40.[i]
Pathophysiology
Essentially, Wilson’s disease results in copper deposition in various tissues, which is what manifests in signs and symptoms. Most commonly, the disease will affect the liver and brain, particularly the basal ganglia.
The gene ATP7B on chromosome 13 is the main culprit in Wilson's disease. There is a mutation in this gene, resulting in disrupted copper excretory mechanisms and a build-up of copper in the body.[ii] In high levels, copper can induce oxidative stress thereby leading to destruction of cells. [iii] This happens in the following ways:
- ATP7B codes for the copper-transporting ATPase 2 protein which is responsible for transferring copper into the bile for excretion, resulting in copper build up.
- Apoceruloplasmin is a serum protein which is responsible for carrying copper around the body. ATP7B is required to incorporate copper into apoceruloplasmin to form ceruloplasmin. Thus, a mutation in this gene, as seen in Wilson’s disease, leads to reduced circulating ceruloplasmin and higher levels of copper.[iv]
Clinical Features
- History of hepatitis: Abdominal pain, jaundice
- Features of cirrhosis/liver failure: Ascites, peripheral oedema, oesophageal varices, bruising, fatigue, spider naevi etc.
- Neurological symptoms:
- Tremor
- Dysarthria
- Ataxia
- Dysphagia
- Parkinsonism: From copper deposition in the basal ganglia
- Psychiatric Manifestations: Depression, mania, personality changes, psychosis
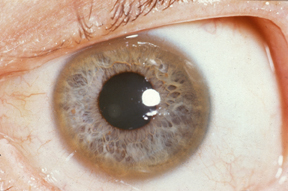
- Copper deposits also occur in the Desçemet’s membrane of the cornea, forming Kayser-Fleischer rings which are visible through slit-lamp examination.[v]
Herbert L. Fred, MD, Hendrik A. van Dijk, CC BY 3.0 , via Wikimedia Commons
Kayser-Fleischer Rings
Investigations
Bedside and Examination
- Slit-lamp examination: Kayser-Fleischer rings
- 24-hour urinary copper: High
Bloods
- Liver function test: This is performed to look for an abnormality. Patients may have poor synthetic function of the liver e.g. low albumin if there is liver failure
- Serum ceruloplasmin: Low. Wilson’s disease is a mutation in the gene ATP7B which is responsible for binding copper to apoceruloplasmin to form ceruloplasmin, a protein which carries copper in serum. Mutated ATP7B leads to low levels of ceruloplasmin.
- FBC, U&E, Clotting screen: Baseline
Imaging
MRI brain: Looking for degenerative changes[vi]
Special Tests
- Liver biopsy: Liver copper present and there may be signs of cirrhosis or hepatitis – this is the most definitive method of diagnosis.
- Genetic testing: Looking for ATP7B mutations
Management
Conservative
Avoiding foods high in copper such as chocolate, mushrooms and nuts, is recommended. Patients should also avoid hepatotoxic substances such as alcohol and hepatotoxic drugs.
Pharmacological
Copper chelating agents are used in order to reduce the levels of copper in the body. Penicillamine is typically used as the first-line drug although other medications such as trientine are also used.
Zinc acetate prevents the absorption of copper rather than acting as a chelating agent.[vii]
References
[i] https://www.ncbi.nlm.nih.gov/books/NBK441990/
[ii] https://ghr.nlm.nih.gov/gene/ATP7B
[iii] https://www.ncbi.nlm.nih.gov/pubmed/15554419
[iv] http://www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/hepatology/wilson-disease/#pathophysiology
[v] https://www.spg.pt/wp-content/uploads/2015/11/2012-wilson-easl.pdf
[vi] Oxford Handbook
[vii] https://britishlivertrust.org.uk/information-and-support/living-with-a-liver-condition/liver-conditions/wilsons-disease/